Our Research
Cancer is considered to be a global health problem. The incidence and mortality rate of cancer is steadily increasing worldwide. Moreover, the increasing resistance to the established anticancer drugs is of grave concern.
Protein kinases as the main therapeutic targets:
Protein kinases play a very significant role in various cellular functions such as cell signaling, cell differentiation, and cell proliferation. Deregulation of the protein kinase activity has emerged as a major mechanism by which cancer cells evade normal physiological constraints on growth and survival. Such aberrant functions of the kinases in a cancer cell have highlighted them as one of the most successful families of drug targets. Our research group at IITGN focusses on the chemical biology of cancer-related protein kinases involved in the DNA damage response pathways which can be used as targets for the anti-cancer therapy. We use small molecule inhibitors approach to study these proteins so as to develop new age cancer therapeutics.
- H.pylori infection - Stomach Cancer
- DNA Damage Sensing kinases
- Targeting RAS GTPase
- Validation of MDC1
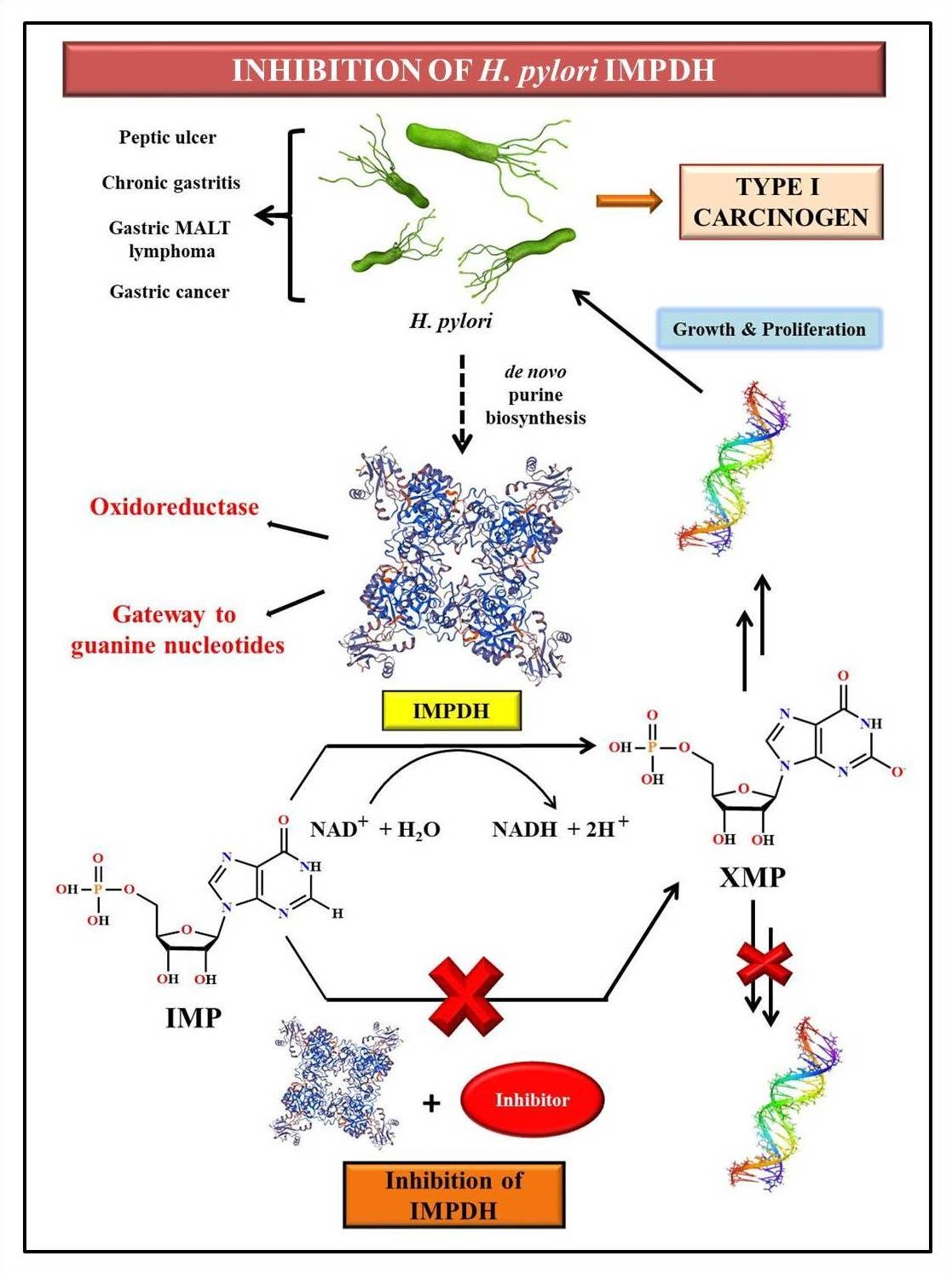
Helicobacter pylori (H. pylori), a member of the 𝜺 subdivision of Proteobacteria, is a gram-negative spiral-shaped bacterium, and a well-known gastric colonizer. Worldwide, H. pylori infection affects almost 50% of the population, with the developing countries being affected the most. The clinical features of the infection include upper gastrointestinal disorders such as peptic ulcer, chronic gastritis, gastric mucosa-associated lymphoid tissue (MALT) lymphoma, and gastric cancer. As a result, H. pylori is identified as a Type I carcinogen. In addition, the extensive genetic heterogeneity shown by these species has resulted in the increased antibiotic resistance of several H. pylori strains. Hence, it is essential to identify novel drug targets to treat the infection. Inosine-5’-monophosphate dehydrogenase (IMPDH), an oxidoreductase enzyme involved in the de novo purine biosynthesis, has been recently identified as a potential drug target due to its vital role in bacterial growth and proliferation. The oxidation of inosine-5’-monophosphate (IMP) to xanthosine-5’-monophosphate (XMP) is catalyzed by IMPDH, resulting in the generation of guanine nucleotides which in turn serve as precursors for RNA and DNA. In fact, the inhibition of the bacterial IMPDH would result in depletion of the guanine nucleotide pool, hence hindering the growth and proliferation of H.pylori in the host. Moreover, studies that indicate the amplification of IMPDH in tumors and rapidly proliferating tissues establish the promising future of IMPDH as an anticancer target. The lab focuses on the design and synthesis of potential inhibitors of H. pylori IMPDH as a means to cure gastrointestinal disorders and in the prevention of gastric cancer in the long term. In addition, the effective combination of organic synthesis, molecular biology, in silico studies, and crystallization techniques nurture the enzyme inhibition studies, thus paving the way for path-breaking research in anticancer therapy.
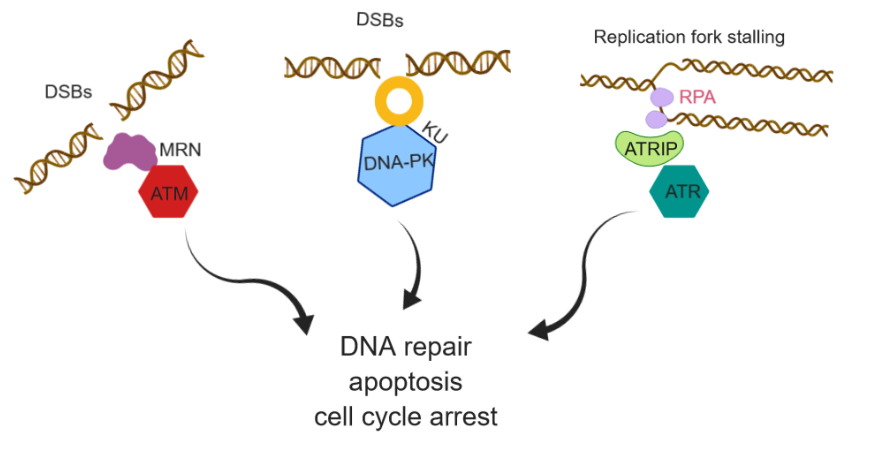
Failure of cytotoxicity of DNA damage-inducing regimes such as radio- and chemotherapy in cancer treatment is mainly due to activation of DNA damage and response (DDR) pathway. DDR pathway is a complex network of the cellular process that maintains genomic stability in the normal cell. Defects in the DDR pathway in a cancer cell often predispose the mutagenic repair pathway that drives oncogenesis. Sensitizing cancer cells selectively through targeting enzymes in the DDR pathway in combination with radio- and chemotherapy in individual cancer cell types can bring synthetic lethality and also cancer-specific vulnerability. Developing specific DDR inhibitors has remained a challenging task over the last few decades with significantly fewer drugs entered in clinical trials.
ATR Kinase
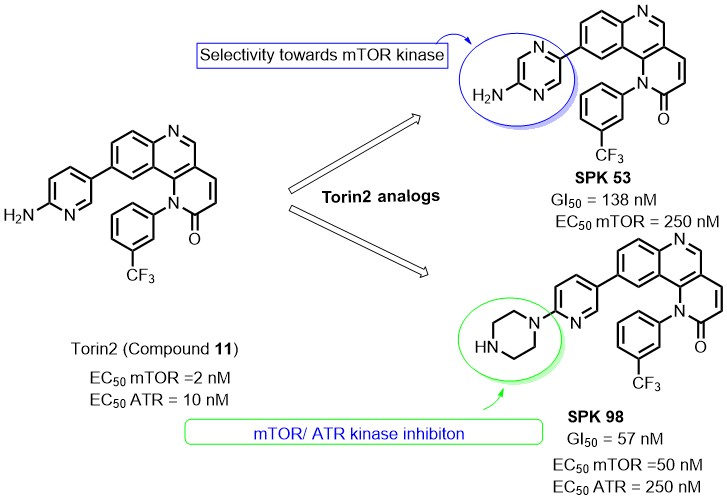
ATR gets activated in response to single-strand DNA breaks caused by UV irradiation/ chemotherapy. Activation of ATR kinase leads to the phosphorylation of several cell cycle checkpoint kinases to arrest the cell cycle for the repair of damaged DNA. By targeting ATR kinase along with radiotherapy/chemotherapy, one can achieve the selective targeting of cancer cells over normal cells. Also, this combinatorial approach proved to have reduced toxicity (organ failure) and resistance caused by drugs targeting DNA replication machinery. Torin 2 is an ATP competitive mTOR inhibitor (EC50 = 2nM ) also known to inhibit other PIKK family kinases (ATR, DNA-PKcs, ATM). As part of our kinase drug discovery research, we have designed few Torin2 analogs and synthesized them through multistep synthesis. All the synthesized compounds were characterized by NMR and mass analysis. The newly synthesized analogs were evaluated for their anti-cancer activity via CellTiter-Glo®assay. Additionally, compounds SPK 98 and SPK 53 showed significant inhibition for ATR and mTOR substrates, i.e., p-Chk1 Ser 317 and p70 S6K Thr 389, respectively. Compounds SPK 98 and SPK 53 displayed promising anti-cancer activity with HCT-116 cell lines in the preliminary assay and looking further to perform preclinical studies.
ATM Kinase
The project aims at inhibiting ATM kinase, which arbitrates the repair of the DNA double-strand breaks. Creating genomic instability by causing DNA strand breaks is used as a mode of treatment (certain chemotherapeutics and radiotherapy) for cancer. Due to the elevated activity of DNA Damage and Response (DDR) pathway in cancer cells, such treatments face resistance. Therefore, it is of utmost importance to address this issue. We have designed and synthesized quinoline-3- carboxamide derivatives as ATM kinase inhibitors. The synthesised quinoline-3-carboxamide were tested against various cancer cell lines and showed significant toxicity. The compounds were also 2-3 fold less toxic to normal cell line (293T). The synthesized compounds will further be modified to enhance their toxicity against cancer cells.

TLK1B Kinase
Tousled like kinases (TLK) have been implicated in chromatin assembly, DNA repair, DNA replication, and transcription. They are expressed in most cell lines with the highest activity levels in the S-phase of the cell cycle. TLK belongs to a class of serine-threonine kinases that phosphorylates important DDR proteins like ASF1, RAD 9, NEK 1. TLK activity is attenuated during the DDR activation via the ATM and Chk 1 kinase pathway, as the Chk 1 directly phosphorylates its C-terminal residue. This suggests that TLK can be used as a target to hinder the DDR pathway specific to cancer cells. Our project mainly focuses on targeting TLK 1 for the inhibition of DDR. We have designed and synthesized phenothiazine based derivatives as potential TLK inhibitors, which showed promising results in in vitro and in vivo studies. Further, we also aim at modifying these derivatives and exploring novel scaffolds for the target using in-silico studies, to achieve a better inhibition towards TLK during biological studies.
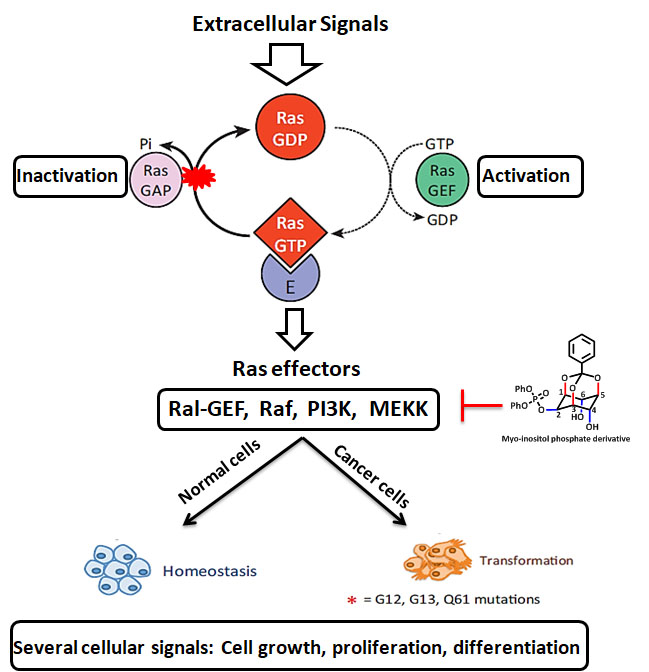
RAS GTPases
RAS mutations are known to be the most recurrent gain-of-function changes instigated in patients with cancer. The RAS gene family is often mutated in most of the human cancers (9-30%) and the pursuit of inhibitors that bind to mutant RAS continues as a foremost target. Ras is a small family of GTPases that control numerous cellular functions like cell proliferation, growth, survival, gene expression, and is closely engaged in cancer pathogenesis. There are no specific molecules reported targeting the same, although it is a known as oncogene for more than three decades. In our recent study, new phosphate derivatives of Myo-inositol are designed and synthesized to inhibit the oncogenic KRAS pathway in breast cancer cells, which has been validated by cellular and theoretical studies. Our inspiration for designing potential RAS inhibitors was based on the SML-8-73-1(first covalent inhibitor targeting KRAS G12C) to overcome its existing limitations such as cell impermeability. It could offer a base for scientific and clinical development of further modified or improved form of Myo-inositol phosphate derivatives for the development of targeted therapy against oncogenic KRAS, which may contribute to the future study for breast cancer.
Validation of MDC1 as a therapeutic target
Cisplatin, the most common chemotherapeutic drug for the treatment of advanced-stage cervical cancers has limitations in terms of drugs resistance observed in patients partly due to functional DNA damage repair (DDR) processes in the cell. Mediator of DNA damage checkpoint 1 (MDC1) is an important protein in the Ataxia telangiectasia mutated (ATM) mediated double-stranded DNA break (DSB) repair pathway. In this regard, we investigated the efect of MDC1 change in expression on the cisplatin sensitivity in cervical cancer cells.
Results: Through modulation of MDC1 expression in the cervical cancer cell lines; Hela, SiHa, and Caski, we found that all the three cell lines silenced for MDC1 exhibited higher sensitivity to cisplatin treatment with inefficiency in accumulation of p γH2AX, Ser 139 foci, and increased accumulation of pChk2 Thr 68 at the damaged chromatin followed by enhanced apoptosis. Further, we observed the increased p53 Ser 15 phosphorylation in the MDC1 depleted cells. Our studies suggest that MDC1 expression could be a key determinant in cervical cancer prognosis and its depletion in combination with cisplatin has the potential to be explored for the sensitization of chemo-resistant cervical cancer cells.